
Myambutol
Discount myambutol 600mg line
Genetic testing is also just beginning to be used to inform prescribing of drug therapy based on individual genetic variation (pharmacogenetics) virus 5 hari cheap 600 mg myambutol with visa. In 2006, testing is offered internationally, through both public and private sector genetic testing services, and there is evidence that human samples and related data are being exchanged across borders in an environment where regulatory and oversight procedures vary significantly between jurisdictions. It also documented existing quality assurance practices in use in molecular genetic testing laboratories and policies for the handling of samples and genetic data including transfers across borders. The survey confirmed the steady growth of molecular genetic testing and its widespread availability. The survey also showed that laboratories in all countries use both formal and informal professional referral networks that exist either within or outside each country to send samples across borders. Some countries have well established licensing, accreditation and certification procedures to 1. One reason for this could be that regulations with which laboratories must comply are not specifically designed for molecular genetic testing. Considerable differences exist in the use of licensing, certification, and accreditation procedures and this poses a number of challenges for molecular genetic testing, particularly with respect to the standards under which tests are performed and results are reported for clinical application, and the training and qualifications required by laboratory personnel. Consequently, there is uncertainty about terminology and the choice of the most appropriate quality system. There is also a lack of understanding amongst the international community on the mutual acceptability of quality assurance systems. As laboratories increasingly provide their services to both national and international customers there is a need to develop international consensus and best practice to assure consistency in the quality of services available. The Principles are policy recommendations specifically directed to Governments and those involved in the regulation of genetic services. Best Practices are recommendations that aim to provide operational guidance in implementing the Principles and are directed to professional bodies and providers of molecular genetic testing services in developed and developing economies. They are relevant to healthy people as well as those showing symptoms of a condition and may have important implications for the relatives of the person tested. The single laboratory test to establish a genotype is usually not repeated and its result forms a permanent part of the medical record. Consequently, it is important that services are provided with the appropriate level of support to the patient and their family prior to the offer of a genetic test and following the result. Whilst good laboratory practices and adherence to quality standards are the responsibility of all medical testing laboratories, these features of molecular genetic testing place an enhanced duty on laboratories to assure the quality of their services. Research laboratories play a valuable role in the development and validation of new tests particularly in the provision of genetic testing for rare diseases. Governments, regulators and professional bodies have a responsibility to ensure that all genetic testing services are offered within a quality assurance framework that retains the confidence of the public. These Guidelines comprise Principles and Best Practices for quality assurance in molecular genetic testing for clinical purposes. The ethical and legal principles set out in international declarations and agreements and the diversity of systems and jurisdictions within and between countries have been recognised during the development of these Guidelines. They concern molecular genetic testing offered in a clinical context, and the quality assurance practices of laboratories that carry out such tests. They focus on molecular genetic testing for the diagnosis of a particular disease or condition and predictive genetic testing often carried out before any clinical signs of the disease or condition appear. Molecular genetic tests require particular consideration since these tests may be performed on asymptomatic individuals and results may have relevance to important lifetime decisions both for the individuals being tested and for their family and children. The Guidelines reflect this particular responsibility of molecular genetic testing and place emphasis on the accuracy of all aspects of the testing and reporting process including forming links to appropriate levels of counselling. In part, these Guidelines are also relevant and applicable to aspects of clinical cytogenetics testing and biochemical genetic testing. They are not designed to address directly the areas of testing for somatic mutations, variants important in tissue matching, genetic analysis of pathogenic organisms and identity testing, though all share related technologies. These Guidelines are intended to be evolutionary in nature and will therefore need to be reviewed within four years of adoption and thereafter periodically in light of new genetic knowledge, technological advances, evolution of quality management and societal needs and to ensure that they are achieving the desired objectives. The Principles provide a framework within which to conceive measures that assure all aspects of quality, including competence to carry out and report molecular genetic tests as well as the education and training of laboratory personnel. Part Two of the Guidelines contains explanatory Annotations which elaborate on the Principles and Best Practices in Part One. Finally, a glossary of terms as well as a list of other relevant publications is provided. Scope this Recommendation applies to quality assurance of molecular genetic testing offered in a clinical context. It focuses on molecular genetic testing for the diagnosis of a particular disease or condition and predictive genetic testing often carried out before any clinical signs of the disease or condition appear. Molecular genetic testing should be delivered within the framework of health care. It should be proportionate and appropriate to the characteristics of the test, the test limitations, the potential for harm, and the relevance of test results to individuals and their relatives. All molecular genetic testing results for clinical care purposes should be reported by competent laboratories, as established by accreditation or other equivalent recognition consistent with these Guidelines. Research laboratories carrying out molecular genetic testing which are not accredited nor hold an equivalent recognition should arrange for such results to be verified and reported by a laboratory holding such an accreditation or recognition. Proficiency testing: monitoring the quality of laboratory performance Principles C. When not available, they should participate in alternative methods relevant to the tests they perform. The indication for testing and specific medical information where it is relevant to test interpretation. The test performed and the methodology used (including the scope of the analysis, the limitations of the test and its analytical sensitivity and specificity). The name and location of laboratory(ies), including any referral laboratory(ies), which performed the actual testing on the sample. An interpretation of the result in the context of the indication for testing and all other information provided to the laboratory. Development of educational and training programmes should be encouraged where they do not exist. Relevant government or professional authorities should recognise medical genetics as a discipline comprising both a clinical and a laboratory specialty. These measures should be comparable to those applied in other areas of laboratory medicine. They should include systems to validate requirements for education, training, qualifications and skills specific to molecular genetic testing. The purpose of these Annotations is to provide additional information on the Principles and Best Practices found in Part One of the Guidelines. For ease of reference, Principles or Best Practices to which a specific annotation alludes to are included in parentheses at the end of the relevant paragraph. These Guidelines offer principles and best practices for quality assurance in molecular genetic testing for clinical purposes. They are addressed to all those involved in the regulation and provision of molecular genetic testing. The Guidelines recognise the existence of regional, national and international quality assurance frameworks and seek to facilitate their mutual recognition. This mutual recognition can only be achieved through international consensus on minimum common standards to assure consistency in the quality of molecular genetic testing services. It has been suggested that many factors may contribute to this, including uncertainty about terminology and the choice of the most appropriate quality system. Instruments relevant to quality assurance may include: accreditation, licensing, certification, proficiency testing, internal quality control measures, documentation of policies and procedures, and/or assurance of personnel competence that may include certification or registration of laboratory personnel. Accreditation is a procedure by which an authoritative body gives 4 formal recognition that a body is competent to carry out specific tasks. It is only granted after a thorough on-site assessment by technical assessors of the management, environment, policies and procedures of the laboratory in addition to specific scientific/technical competences measured against external standards. International standards are relevant to the design of jurisdiction specific accreditation systems. These standards are not themselves accreditation systems but may be referred to by the authoritative bodies that award accreditation. Accreditation standards related to clinical laboratories place emphasis on having an effective quality assurance system in place; on a commitment to meeting the needs of patients and their doctors as users of laboratory services; and on a need for a continuous cycle of quality improvement at the centre of all policy making and operational decisions.
Purchase 400 mg myambutol amex
Now pipistrel virus myambutol 600 mg with mastercard, it is believed that there are two patho-genetically distinct pathways for the development of colon cancer involving stepwise accumulation of multiple mutations. However, the genes involved and the mechanisms by which the mutations arisen are different. The molecular evolution of colon cancer along this pathway occurs through a series of morphologically identifiable stages. This is followed by the formation of small adenomas that progressively enlarge, become more dysplastic, and ultimately develop into invasive cancers. Loss of this gene is believed to be the earliest event in the formation of adenomas. There are overwhelming genetic data to support the role of E-cadherin as a tumor/ invasion suppressor in epithelial cells,and loss of expression, as well as mutations, has been described in a number of epithelial cancers. The genetic studies up to the present are sustaining the suppressor invasive/tumoral role of E-cadenin in the epithelial cells, and the expression loss along with mutations were described in some types 94 Mutations in Human Genetic Disease of epithelial cancers (breast, colorectal, thyroid, endometrium, ovary cancer). It is presumed that the loss of function contributes to the cancer progression by increasing the level of proliferation, invasion and/or metastasis. E-cadenin expression modifications are frequently associated with a high tumoral level, like the disease of prostate, breast, bladder, pancreas, stomach and colon. As an epithelial cell adhesion molecule E-cadherin mediates the contact between neighboring epithelial cells, including the colorectal epithelial cells, and helps to establish the defined membrane domains and cell polarity (Goodwin and Yap 2004). The extracellular domain of E-cadherin is responsible for homotypic binding of adjacent cells, and the cytoplasmic domain of E-cadherin facilitates adhesion through interaction with catenin proteins (Bryant and Stow 2004). The ectodomain of this protein mediates bacterial adhesion to mammalian cells and the cytoplasmic domain is required for internalization. E-cadherin expression in epithelial cells is crucial for the establishment and maintenance of epithelial cell polarity. Mutations at the level of this gene are responsible in part for inherited predisposition to ovary, breast, prostate and colon cancers. However, whether these mutations are a factor in sporadic forms of these tumours remains unclear. Mutations in this gene are responsible in part for the inherited predisposition to breast and ovarian cancers, and probably for one third of all site specific inherited breast cancer. Double-strand breaks in mammalian chromosomes stimulate the activity of recombination repair enzymes by more than 100-fold. The plasma concentration of the enzyme increases dramatically in severe infections and other diseases involving generalized infiammation and cancer (Ogawa M, 1991). On the one hand, it has been suggested that a mutation resulting in splice variants of the Pla2g2a gene and in different truncated forms of its protein accounts for the increased number of polyps in mice carrying the Min mutation. The variability in clinical presentation, aggressiveness, and patterns of treatment failure suggests distinct genotypes and phenotypes identification, which can help future treatment strategies. There is an increasing interest in this therapeutic strategy on the part of pharmaceutical and bio-pharmaceutical companies, consumers, and third party payers. Consequently, the level of clinical trial activity surrounding personalized medicines is intensifying as sponsors seek ways to target their therapies to patient populations that would most benefit from them. The idea of applying such a model to our studies was generated during the research that we conducted in our projects. We have noticed that between different proteins and genes is a very close relationship, which depends on the tumor type, cell grade and staging. Following a study of a large number of articles published in the international databases we observed that other researchers have drawn the same conclusion. Tissue samples and blood Samples were obtained with the consent of 93 patients, consisting of histopatologically confirmed colorectal adenomas. Samples were obtained during colonoscopy with biopsy forceps, by harvesting at least four fragments from all the quadrants of the pathological tissue. After surgical resection, tumor tissues were cut in small pieces, frozen immediately in liquid nitrogen and stored at 800C until they were analyzed. For the initial patients group, only 75 patients who had at least 75% tumor cells were taken in consideration for molecular biology analyses. Clinicopathological characteristics the medical records of all 93 patients provided their birth date and sex, and the following parameters: tumor location, tumor size, lymph node metastases, pathological stage, vascular and neural invasion and tumoral differentiation grading. Our study has not taken into consideration the diet, because most of the patients do not know the food properties or they use food with pro-carcinogen potential. Regarding the diet, we consider that the patient instruction is extremely useful and has to be done by the surgeon doctor after the surgical treatment and then by the family doctor. This approach allows both secondary prophylaxis and control of possible relapses/ recidivists. A monitoring of the patients included in the study will shows the efficiency of medical control 98 Mutations in Human Genetic Disease and the conscious of this mortal disease. In the studied lot of patients we have not registered cases with relapse, and we cannot predict their future behavior. Immunohistochemical expression by immunofluoresce of the studied proteins Because the interpretation of immunohistochemistry analyses remains the basic of anatomic pathology, in our study we first evaluated the protein expression of the key point proteins that were taken in our study. Unlike the normal histopathological analyses, our evaluation was based on protein fluorescent signal which, from our point of view, is more specific than classical immunohistochemistry. It is one of the four muscle actin isoforms, a protein involved in supporting basic contractile apparatus in muscle cells. This expression can be found in vascular cells, intestinal muscularis mucosae and muscularis propria, and in the stromal tissue. In normal tissue, the immunofluorescence signal is strong (+3) around tumor crypts, in the vessel walls and stromal smooth muscle Genotype-Phenotype Disturbances of Some Biomarkers in Colorectal Cancer 99 fibers. Smooth muscle, used as a positive marker for immunofluorescence signal, have immunofluorescent signal in blood vessels, intestinal muscularis mucosae and muscularis propria, and in the stromal tissue. On section obtained from patient 3 we can observe a weak intensity on the apical part of epithelial cells and loss of signal, too. In the apical half of the fluorescent signal crypt, epithelial cells and infiltrated cells disappeared (-). In normal colorectal tissue, fi-catenin expression appears on the membrane of epithelial cells. A normal expression with immunofluorescent signal on cytoplasm and on the border of crypts can be observed on the section from patient 8. On section from patient 3 we can observe an over-expression in the cytoplasm/ nucleus of epithelial cells and loss of expression in the membrane. We can observe how the fluorescent signal on the membrane of epithelial cells gradually decreases in intensity during the tumor progression, along with increased fluorescent signal by over-expression in cytoplasm (in 28 patients) and in the nucleus (in 5 patients). Regarding E-cadherin expression, colorectal tumors showed a heterogeneous type of expression compared to the normal colorectal epithelium in which E-cadherin expression is present on the basolateral membrane to the whole length of the glandular crypts and on the intercellular membranes. In the case of lymph nodes analyses, Genotype-Phenotype Disturbances of Some Biomarkers in Colorectal Cancer 101 Figure 4. A normal expression with immunofluorescent signal on the membrane of epithelial cells can be observed on section from patient 70. In the case of patient 74 we can observe a reduced/ loss of expression in the epithelial cell membranes. On patient 73 an over-expression in the cytoplasm of epithelial cells and in some infiltrating cells was noticed. On patient 60 we can observe an over-expression on the epithelial cells from the crypt foci. On other sections from patient 60 over-expression was observed only on the apical pole of epithelial cells. On patient 43 we can observe an over-expression on the membrane of epithelial cells from the crypt foci. It is also a useful tool for the diagnosis of genetic diseases characterized by large genomic rearrangements.
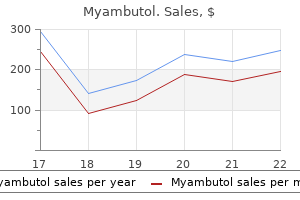
Myambutol 600mg on line
Assess intellectual/cognitive abilities with special attention for learning difficulties as a result of motor delay antibiotics for uti trimethoprim buy myambutol 800mg mastercard, executive dysfunctions and inattention. Ongoing review and support of learning and development with further assessment of special educational needs as required. Persistent vomiting or food refusal may require tube feeding (although this is rare). Enrol patient in an individualised preventative oral healthcare programme from an early age. Routine follow up and regular dental examinations by a family dentist or local community dental services are essential. Maternal considerations Potential difficulties, for example those arising from coagulation defects during childbirth, should be considered and planned for as appropriate. Previously diagnosed adults: regular cardiac assessment of existing heart disease, or cardiac evaluation incase aortic disease missed previously. A case with a platelet cyclooxygenase-like deficiency and chronic idiopathic thrombocytopenic purpura. It contains information on over 5,000 conditions, including Williams Syndrome, and lists specialised clinics, diagnostic tests, patient organisations, research projects, clinical trials and patient registries relating specifically to Noonan Syndrome. This new approach uses individual budgets and direct payments to allow individuals more choice and control over the support they receive. Based on a review of the neurology of dyslexia, the model specifies that: 1) Genetically determined focal cortical anomalies in specific left perisylvian language areas are the underlying cause of the phonological deficit; 2) this phonological deficit is the primary cause of reading impairment; 3) Under certain hormonal conditions during gestation, these cortical anomalies induce secondary disruption in sensory pathways, notably in the thalamus. The disruption may even extend to further areas, like the posterior parietal cortex and even the cerebellum; 4) When this happens, the individual affected displays one or several components of a sensorimotor syndrome, which may in some cases aggravate the reading impairment. The model generalises to specific language impairment and possibly to other domain-specific developmental disorders, each particular disorder being characterised by the specific location of the brain anomalies. Certain theoreticians consider them to be domain-specific disorders, arising from congenital dysfunctions circumscribed to certain cognitive components. Others think that these disorders are much more general, and that the seemingly specific components affected are in fact part of a more extended syndrome, usually encompassing the sensory and motor domains (Stein and Walsh 1997; Karmiloff-Smith 1998; Tomblin and Pandich 1999; Gepner and Mestre 2002). Some of these researchers even hold that domain-specific developmental disorders are, in principle, unlikely to exist at all (Thomas and Karmiloff-Smith 2002). In the case of developmental dyslexia, the predominant theory is that it is due to a specific phonological deficit (Snowling 2000). Nevertheless, this view has been challenged by increasing evidence of sensory and motor disorders in dyslexics, leading to competing theories implicating auditory/temporal processing deficits (Tallal 1980; Farmer and Klein 1995), visual/magnocellular dysfunction (Lovegrove et al. In the face of this highly diverse and inconsistent data set, only one theory so far has attempted to account for all the empirical evidence: the general magnocellular theory, in which a generalised dysfunction of magno-cells affects all sensory pathways and further spreads to the posterior parietal cortex and the cerebellum, thereby encompassing all the known cognitive, sensory, and motor manifestations of dyslexia (Stein and Walsh 1997; Stein 2001). In particular, it fails to explain why the prevalence of sensorimotor dysfunction is so much lower than that of the phonological deficit in the dyslexic population. Even within the subset of dyslexics affected by sensory and/or motor disorders, the causal relationship with the reading impairment is far from clear (Ramus 2003; Rosen 2003). On the basis of a comprehensive review of the literature, I have previously advocated that dyslexia is, in most individuals, explained by a specific phonological deficit; furthermore, a more general sensorimotor syndrome occurs more often in the dyslexic than in the general population, but does not by itself play a causal role in the aetiology of the reading impairment (Ramus 2003). According to this view, a complete theory of dyslexia must explain both how a specific phonological deficit might arise, and why a sensorimotor syndrome should be significantly associated with it. Specifically, it potentially explains how a phonological deficit may arise from genetically determined brain anomalies, in isolation in certain individuals, or in conjunction with sensorimotor impairments in others. This model is compatible with all the known genetic, neurological, and cognitive data available on dyslexia. It further suggests explanations for a few puzzling issues like co-morbidity between and heterogeneity within disorders, and makes a number of specific predictions yet to be tested. Insights from anatomical studies and animal models Post-mortem examination and brain imaging studies have documented many differences between dyslexic and control brains, in the left peri-sylvian cortex (Galaburda et al. In most cases, the functional significance of these brain differences has not been elucidated. It is not even clear which of those differences are specifically relevant to dyslexia, considering the well-known comorbidity between dyslexia and many other disorders (Kadesjo and Gillberg 2001; Kaplan et al. Nevertheless, the functional significance of two types of brain anomalies has been studied in greater detail. Anomalies of cell migration called molecular layer ectopias and focal microgyri have been observed by Galaburda and colleagues in the peri-sylvian cortex of dyslexic brains (Galaburda and Kemper 1979; Galaburda et al. Ectopias consist of 50-100 neurons and glia that have escaped into the molecular layer of the cortex through a breach in the external glial limiting membrane, accompanied by mild disorganization of the subjacent cortical layers. Microgyria are more severe disturbances where the organisation of all layers of the cortex is severely affected. It is quite natural to hypothesise that anomalies in the magnocellular layers of the lateral geniculate are the cause of visual deficits, and that anomalies in the medial geniculate are the cause of auditory deficits. Similarly, it is easy to see cortical anomalies in left peri-sylvian areas as the underlying cause of phonological, and perhaps other cognitive difficulties. In this anatomical evidence, one can therefore see direct neurological support for auditory and magnocellular theories of dyslexia. The implicit causal (bottom-up) scenario is that anomalies in the thalamus engender ectopias and microgyria in certain cortical areas to which the thalamus is connected. At the cognitive level, this would translate into the auditory deficit causing a phonological deficit, and into the basic visual deficit causing visual attention/planning problems, as prescribed by the magnocellular theory. Indeed, Galaburda and colleagues have found that, at least in animal models, the causal direction seems to be the opposite (top-down), i. Indeed, it is possible to surgically induce ectopias and microgyria by poking a hole in the external glial limiting membrane of the developing cortex of rats during late neocortical neuronal migration. There are also strains of mutant mice that spontaneously develop similar malformations. This suggests that the direction of causation is indeed top-down, from the cortex to sensory relays in the thalamus. Similar auditory disorders are found in ectopic mice, regardless of the localisation of ectopias (Peiffer et al. Another interesting aspect uncovered in these studies is that only male rats were initially found to have impaired auditory function following early inducement of microgyria (Fitch et al. Similarly, only male ectopic mice show auditory deficits (Peiffer, Rosen, and Fitch 2002). Finally, the cortical anomalies themselves seem to have an impact on cognitive function: ectopic mice and rats with spontaneous or induced ectopias and microgyria exhibit a variety of learning deficits (Denenberg et al. Furthermore, the location of the cortical disruption influences the specific type of learning deficit exhibited by the animal (Hyde et al. To summarise, these results suggest that, in animal models at least, (1) cortical anomalies (microgyria, ectopias) induce secondary anomalies in sensory relays in the thalamus, but (2) only under certain ffital hormonal conditions. Obviously, there are many more conceivable neuro-developmental models of dyslexia than the one most directly suggested by these particular neurological observations and animal models. But, limited as these data are, they seem more compatible with the idea of a specific phonological deficit optionally associated with additional sensorimotor disorders, than with any theory requiring causation of the phonological deficit through other sensory/cognitive disturbances. I will now spell out and discuss in further detail what a plausible neurological model of dyslexia and other developmental disorders might be, based on this reinterpretation of the anatomical and animal data. It should be emphasised that this model is largely speculative; it attempts to be compatible with all the available data, but given that the available data is not excessively constraining, alternative models are perfectly viable. The goal here is mainly to provide a plausible, testable model that makes specific predictions. A neurological model of dyslexia Focal anomalies and the phonological deficit the main claim of the model is that congenital anomalies in specific left peri-sylvian areas are the direct cause of a phonological deficit, which itself is the direct cause of reading impairment. A simple version of this model attributes the main responsibility to cortical ectopias and microgyria. This is indeed where the main brain areas involved in phonology seem to be located: mainly the supramarginal and angular gyri, the posterior superior temporal gyrus, the insula, and the inferior frontal gyrus, although there is debate as to which areas are involved specifically in phonological representations, and which are more concerned with reading or speaking (Paulesu et al. Note that this does not exclude that areas which become more specifically dedicated to reading (like the left fusiform gyrus. More generally, the multiplicity of areas involved in phonology and reading, together with the multiple differences found between dyslexic and control brains, makes it plausible that several different patterns of cortical disruption will lead to a reading impairment; this diversity may actually underlie the various manifestations of the phonological deficit in dyslexia.
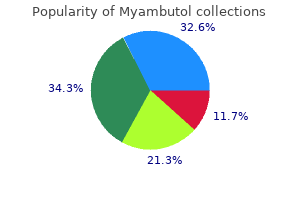
Buy myambutol overnight delivery
These molecules identify which copy of a gene was inherited from the mother and which was inherited from the father antibiotic resistance reversal purchase 800 mg myambutol with mastercard. The addition and removal of methyl groups can be used to control the activity of genes. They do know that imprinted genes tend to cluster together in the same regions of chromosomes. Two major clusters of imprinted genes have been identified in humans, one on the short (p) arm of chromosome 11 (at position 11p15) and another on the long (q) arm of chromosome 15 (in the region 15q11 to 15q13). This loss of gene function can lead to delayed development, intellectual disability, or other health problems. The most well-known conditions include Prader-Willi syndrome, which is characterized by uncontrolled eating and obesity, and Angelman syndrome, which causes intellectual disability and impaired speech. Other conditions, such as Beckwith-Wiedemann syndrome (a disorder characterized by accelerated growth and an increased risk of cancerous tumors), are associated with abnormalities of imprinted genes on the short arm of chromosome 11. The University of Utah offers a basic overview of genomic imprinting learn. Additional information about epigenetics, including genomic imprinting Geneimprint, a website about genomic imprinting, provides an introduction to imprinting. An animated tutorial from the University of Miami illustrates how uniparental disomy occurs hihg. Although it is possible to inherit some types of chromosomal abnormalities, most chromosomal disorders (such as Down syndrome and Turner syndrome) are not passed from one generation to the next. These changes are not inherited, but occur as random events during the formation of reproductive cells (eggs and sperm). An error in cell division called nondisjunction results in reproductive cells with an abnormal number of chromosomes. For example, a reproductive cell may accidentally gain or lose one copy of a chromosome. Some changes in chromosome structure can be inherited, while others occur as random accidents during the formation of reproductive cells or in early fetal development. Because the inheritance of these changes can be complex, people concerned about this type of chromosomal abnormality may want to talk with a genetics professional. Some cancer cells also have changes in the number or structure of their chromosomes. Because these changes occur in somatic cells (cells other than eggs and sperm), they cannot be passed from one generation to the next. For more information about how chromosomal changes occur: As part of its fact sheet on chromosome abnormalities, the National Human Genome Research Institute provides a discussion of how chromosome abnormalities happen. The March of Dimes discusses the causes of chromosomal abnormalities in their fact sheet Chromosomal Conditions. Some genetic disorders are more likely to occur among people who trace their ancestry to a particular geographic area. People in an ethnic group often share certain versions of their genes, which have been passed down from common ancestors. If one of these shared genes contains a disease-causing mutation, a particular genetic disorder may be more frequently seen in the group. Examples of genetic conditions that are more common in particular ethnic groups are sickle cell disease, which is more common in people of African, African American, or Mediterranean heritage; and Tay-Sachs disease, which is more likely to occur among people of Ashkenazi (eastern and central European) Jewish or French Canadian ancestry. It is important to note, however, that these disorders can occur in any ethnic group. For more information about genetic disorders that are more common in certain groups: Know Your Genes from the Genetic Disease Foundation offers a list and descriptions of genetic disorders. The Norton & Elaine Sarnoff Center for Jewish Genetics provides information on disorders that occur more frequently in people with Jewish ancestry, including genetic traits that tend to be more common in Ashkenazi Jews Like many other complex on page 51 traits, studies suggest that both genetic and environmental factors play a role. These ridges are also present on the toes, the palms of the hands, and the soles of the feet. Although the basic whorl, arch, and loop patterns may be similar, the details of the patterns are specific to each individual. The ridges begin to develop during the third month of fetal development, and they are fully formed by the sixth month. The function of these ridges is not entirely clear, but they likely increase sensitivity to touch. The basic size, shape, and spacing of dermatoglyphs appear to be influenced by genetic factors. Studies suggest that multiple genes are involved, so the inheritance pattern is not straightforward. Genes that control the development of the various layers of skin, as well as the muscles, fat, and blood vessels underneath the skin, may all play a role in determining the pattern of ridges. The finer details of the patterns of skin ridges are influenced by other factors during fetal development, including the environment inside the womb. Rare diseases characterized by abnormal or absent dermatoglyphs provide some clues as to their genetic basis. For example, a condition known as adermatoglyphia is characterized by an absence of dermatoglyphs, sometimes with other abnormalities of the skin. Although this gene is clearly important for the formation of dermatoglyphs, its role in their development is unclear. Scientific journal articles for further reading Burger B, Fuchs D, Sprecher E, Itin P. The immigration delay disease: adermatoglyphia-inherited absence of epidermal ridges. Lighter eye colors, such as blue and green, are found almost exclusively among people of European ancestry. Most of the genes associated with eye color are involved in the production, transport, or storage of a pigment called melanin. Eye color is directly related to the amount and quality of melanin in the front layers of the iris. People with brown eyes have a large amount of melanin in the iris, while people with blue eyes have much less of this pigment. The P protein therefore plays a crucial role in the amount and quality of melanin that is present in the iris. Less P protein means that less melanin is present in the iris, leading to blue eyes instead of brown in people with a polymorphism in this gene. Researchers used to think that eye color was determined by a single gene and followed a simple inheritance pattern in which brown eyes were dominant to blue eyes. Under this model, it was believed that parents who both had blue eyes could not have a child with brown eyes. Although it is uncommon, parents with blue eyes can page 135 Genetics Home Reference ghr. The inheritance of eye color is more complex than originally suspected because multiple genes are involved.

Diseases
- Febrile seizure
- Renal dysplasia mesomelia radiohumeral fusion
- Juvenile hyaline fibromatosis
- Kashani Strom Utley syndrome
- Hodgkin lymphoma
- Incontinentia pigmenti achromians
- Dysferlinopathy
- Hypospadias familial
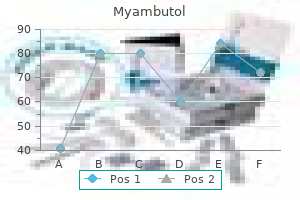
Order 800mg myambutol with visa
M ost recently bacteria glycerol stock buy myambutol 400mg low cost, her laboratory has studied the role of cellular stress responses in the disruption of hematopoietic cell differentiation in myelodysplasia. A second focus of her lab is the role of infammatory cytokines in the anemia of the elderly and in modulating the natural history of myelodysplasia. Carvan held National Institute of Environmental Health Sciences molecular toxicology fellowships at the University of Cincinnati Medical Center. His research uses zebrafsh as a genetic system for identifying genes that infuence the susceptibility of response to xenobiotics. He has been a key participant and architect of the Human Genome, HapM ap, and 1000 Genomes projects. His research focus is the genome-scale analysis of humans and the computational analysis of gene variation and function to understand the molecular genetic basis of complex human phenotypes, particularly disease. He was president of the American Society of Human Genetics in 2008 and received its W illiam Allan Award in 2013. He is one of the founding editors-in-chief of Genome Research and the Annual Reviews of Genomics and Human Genetics, and has served and serves on the boards of numerous international journals, academic societies, the National Institutes of Health, and biotechnology companies. She serves on the editorial boards of the Journal of Nutritional Biochemistry and Epigenetics and is an associate editor for Environmental Health Perspectives, Environmental Epigenetics, and Toxicological Sciences and is an active member of the Society of Toxicology, the Environmental M utagen and Genomics Society, and the American Society for Nutrition, and she served as the chair of the 2015 Gordon Research Conference in M olecular and Cellular M echanisms of Toxicity. Fox began her public health career conducting community health studies around hazardous waste sites as a research scientist in the New York State Department of Health. Until 2007 he was on the faculty of the Harvard School of Public Health and Harvard M edical School. He is interested in the application of laboratory-based biomarkers in chronic-disease epidemiology and tumor biology and in characterizing individual susceptibility to cancer. Her research has focused on the application of biological markers for studying exposures and the interaction between host factors (genetic polymorphisms, nutritional status, microbiome, and epigenetic markers) and environmental exposures. Chan School of Public Health in environmental health and continued her postdoctoral training at Harvard in molecular epidemiology. His research interests are the etiology of cancer and reproductive, perinatal, and pediatric outcomes. Recent work has focused on the role of environmental exposures, genetic factors, and adverse health effects in children and adults. He has also served as a member on four prior committees to review health effects in Vietnam veterans exposed to Agent Orange and other herbicides. Ritz was a research fellow and served her residency at the Psychiatric University Hospital in Hamburg. For the past two decades, she has conducted research on the effects of air pollution on adverse birth outcomes and neurodevelopmental disorders in children who live in Southern California. She also studied the long-term effects of pesticides exposure on Parkinson disease and cancer and is working on establishing a Parkinson disease registry in California. She has been active in Gordon Research Conference programs and was the chairperson for the M echanisms of Toxicology summer session in 2008. In addition to this project, her lab currently uses the zebrafsh model to study the neurotoxicological and behavioral effects following exposure to environmental contaminants during development. She served on the ninth and tenth biennial updates of the Committees to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides. Butler was also a co-editor of Systems Engineering to Improve Traumatic Brain Injury Care in the M ilitary Health System. Over her tenure she has worked on more than 10 studies on a broad range of topics related to the health of military and veteran populations. Styka spent several years working as an epidemiologist for the New M exico Department of Health and the Albuquerque Area Southwest Tribal Epidemiology Center, and she spent several months in Zambia as the epidemiologist on a study of silicosis and other nonmalignant respiratory diseases among copper miners. She has several peerreviewed publications and has contributed to numerous state and national reports. Banks analyzed wellness policies for schools in various counties within Georgia and made recommendations for each county to improve their policy. She is a program offcer within the Division on Earth and Life Studies of the National Academies. Formerly, she was an Environmental Health Scientist at W estat, where she supported the U. Boyle was a student epidemiologist at the M innesota Department of Health and an industrial hygienist at a consulting frm in Cincinnati. She serves as the chair of the nominations committee for the International Society of Exposure Science. She is also a fellow of the Bloomberg American Health Initiative at the Johns Hopkins Bloomberg School of Public Health, where she is pursuing a doctorate of public health in environmental health. Pam ela Ram ey-M cCray, is an administrative assistant in the Health and M edicine Division. She has worked to support numerous studies on military and veterans health, malaria research, and U. Ramey-M cCray worked for the American Psychological Society and the Consumer Product Safety Commission. Reports released in January 2013, January 2014, January 2015, and January 2016 were based on updated scientific literature published since the completion of the 2011 document but maintain the same treatment paradigm. In 2006 and again in 2011 a complete revision was prepared based on published research. Updates of the 2011-revised report were released in January 2013, 2014, 2015, and 2016. The Committee does not make recommendations for therapies that have not been approved by at least one major regulatory agency. Chapter 1 Page 10: the Medical Research Council National Survey of Health and Development recently documented a synergistic interaction between smoking and infant respiratory infection as well as early life home overcrowding with lung function at age 43. Long-term maintenance pulmonary rehabilitation may sustain the benefits achieved after completion of the initial pulmonary rehabilitation program, although one study reported attenuation during follow-up. However, the perception of breathlessness is greater in frequent exacerbators than infrequent exacerbators, (Scioscia et al.
Order cheap myambutol line
For example antimicrobial step 1 order myambutol with a visa, in oncological therapy, organoids of the tumour can be tested for the combination of drugs that eradicates the tumour the most with as few as possible drug-resistant clones. Furthermore, tissues derived in vitro could be generated from patient cells to provide alternative organ replacement strategies. Unlike current organ transplant treatments, such autologous tissues would not suffer from issues of immune-competency and rejection (Lancaster and Knoblich 2014, Ranga, Gjorevski et al. Finally, organoids present the opportunity to bridge the current experimental gap between deep-sequencing efforts in cancer and patient outcome. Currently used preclinical tumour models (cell lines, mouse models) are very limited in their accuracy. As a consequence, many drug candidates that perform well in preclinical models fail to deliver in clinical trials, resulting in suboptimal patient treatment and wasted resources (Sachs and Clevers 2014). Liver organoids, in particular, represent a system with high expectations, particularly for drug testing, because of the unique metabolic profile of the human liver (Lancaster and Knoblich 2014, Ranga, Gjorevski et al. Taken all together, organoids have great potential, yet more research is needed to see whether organoids can truly live up to expectations. Hence, demonstrable evidence of clinicaleffectiveness and cost-effectiveness is urgently needed to support the use of personalized medicine in health care. Here we focus on the most relevant barriers and specific barriers encountered in the Netherlands. Yet, depending on the setting, the confidence of detecting a variant of low allele fraction may be less than it would be had a gene panel been used. Apart from that, the kit used to perform sequencing is a factor to be taken into account, as is the choice of clinical sample, type of data analysis and method to interpret genetic data (Gagan and Van Allen 2015). Because of this, genetic data gathered from different laboratories vary in reliability. This issue was also mentioned by one of the interviewees, who explained that cross validation of sequencing results is by no means common practice in university hospitals. There is as yet no common standard by which one can compare the quality of genomic data between different laboratories. It is therefore of the utmost importance to define internationally accepted definitions of data quality, creating appropriate validation strategies and standards and protocols for proficiency testing for genomics-based tests. It should be noted, as pointed out by one of the interviewees, that market domination of one testing platform from one supplier would be undesirable: assay related errors will only become evident when more platforms are used in practice. Such multidimensional data, where genes can interact with each other or clinical variables, will by necessity require new approaches for their analysis and thought must be given to this when considering the size and design of any clinical study. Additionally, some thought should be given to how we imagine complex genomics data can be interpreted by the clinician and the patient such that clinical management is improved rather than hindered. Matching this single patient against such a dataset would truly enable personalized treatment, but it would need to be visualised in a way that provides clarity for the treating physician and for the patient (McDermott 2015). In addition, this blurring may also require changes in the way health care is financed/ reimbursed. It is described in terms of sensitivity, specificity, positive predictive value and negative predictive value, preferably performed with a prospective randomized controlled trial (Burke 2014). Thirdly, complex pathways are involved in the action and metabolism of most drugs and non-genetic influences also contribute to drug response (Maitland, DiRienzo et al. The diagnostic test criteria sensitivity, specificity and predictive value are applicable to tests for which response is determined as a dichotomous variable. In these situations, the relative contribution of the genotype to the variability in response, i. The clinical importance of the estimated risk depends on the severity of the consequence and is therefore always a personal consideration. As one of the interviewees pointed out, the chance of becoming deaf is of greater significance to a blind person than to someone who is not visually impaired. In general, it can be said that genomic information can be useful, but it is not enough to target a therapy. Moreover, successes in differentiating driver events from passenger events have been moderate, causing response rates to be rather low compared with the number of trials being undertaken. Thus, it is paramount to identify and target the actionable genomic driver events and to differentiate them from passenger events. To address this issue, large-scale sequencing projects and the associated catalogues of somatic mutations are being increased. In addition, temporal and spatial intra-tumour heterogeneity forms a serious problem to genotyping, specifically in cases of oncology. When choosing chemotherapy directed according to a tumour site of origin, a biopsy of any site (either primary or metastasis) can be performed to confirm the diagnosis. But molecular profiling may show dramatic differences in the genotype between the primary site and metastases, and even within different areas of the primary tumour (Greystoke and Chaturvedi 2015). Whether these abnormalities will respond equally to targeted therapy is unclear, but it is unlikely. Equally it is clear that the tumour evolves over time, particularly under the selection pressure of therapy. Selection of drug resistant clones that are probably present at diagnosis occurs rapidly and may even be associated with a change in histology (Greystoke and Chaturvedi 2015). From a clinical standpoint, early detection of resistance is crucial to optimizing therapy, but the way to handle secondary resistance is still unclear; cell eradication seems the only way (Arnedos, Vicier et al. A promising development is the application of organoid technology combined with ultra-deep sequencing in multiple regions of a tumour to determine which drug or combination of drugs would have the highest chance of preventing and/or counteracting resistance. Indeed, targeted therapies have not resulted in major decreases in the number of cancer deaths. In that sense, all kinds of measures, including the traditional public health measures of screening, early detection, and lifestyle changes such as smoking reduction, seem to be necessary to reduce mortality in cancer. Clinical trials Apart from that, measuring clinical validity depends on the types of performance parameters that are chosen. For example, one can measure the penetrance of genetic variation on drug effects through retrospective studies. But one can also measure the effect a certain genetic variation has on an intermediate phenotype, such as drugmetabolizing enzyme activity. Additionally, data can be gathered from in vivo pharmacokinetic or other functional studies, in vitro functional studies, and preclinical and clinical studies that link pharmacological effects or drug concentrations to genetic variation. Due to the small populations described, multiple factors that drive a certain drug effect and the lack of knowledge about this, the results from genetic testing for stratified medicine have been disappointing. Especially in the field of oncology, major attention is being paid to the problem of trials for patient stratification. In cancer, most of the current candidate drivers are detectable in less than 10% of the patients. This approach generates two major issues: firstly, when a patient is tested for a single gene alteration, the likelihood of it being positive and, therefore, treated with a drug matched to a genomic alteration is very low. One solution to address this issue is to use high-throughput genomic approaches to detect all present genomic alterations and enable the patient to be assigned to a specific therapeutic trial. The second issue is the need to screen a large number of patients in order to perform a clinical trial, accrue data and gain enough statistical power. See the reviews of Hollingsworth and Arnedos et al for more information on cancer trials (Arnedos, Vicier et al. In these cases, the gene is not a target of the drug itself, but rather a generic mediator. Many of the interviewees argue for a direct implementation of the found polymorphisms in clinical practice, so that dose adaptations can be made for the medications that are metabolized by the specific gene.
Discount 800 mg myambutol fast delivery
An index of barriers for the implementation of personalized medicine and pharmacogenomics in Europe antibiotics for acne beginning with t myambutol 800 mg without prescription. Accreditation Standards and Key Elements for the Professional Program in Pharmacy Leading to the Doctor of Pharmacy Degree. RxGenomix Collaboration: American Pharmacists Association Co-Provided Pharmacogenomics Training Program. Multiplex genetic testing: Reconsidering utility and informed consent in the era of next-generation sequencing. The Precision Oncology Annual Trend Report: Perspectives From Payers, Oncologists, and Pathologists. Department of Otorhinolaryngology-Head & Neck Surgery, Hospital Universitario Donostia, San Sebastian, Spain. Department of Otorhinolaryngology-Head & Neck Surgery, Hospital Quironsalud Valencia, Spain. Department of otolaryngology-Hospital Complex of Santiago de Compostela, Santiago de Compostela, Spain. Division of Phoniatrics and Audiology, Department of Mental and Physical Health and Preventive Medicine, University of L. Department of Otolaryngology-Head & Neck Surgery, Morgagni Pierantoni Hospital, Forli, Italy. Funding: Compliance with Ethical Standards Disclosure of potential conflicts of interest: Authors have no conflict of interest. Informed consent: Patients were invited to participate and the informed consent was obtained Conflict of interest statement: the authors have no conflicts of interest Correspondence to: Dr. The following epidemiological and clinical outcomes have been studied: age, sex, ethnicity, comorbidities, general and otolaryngological symptoms. The most prevalent general symptoms consisted of cough, myalgia and loss of appetite. Face pain and nasal obstruction were the most specific otolaryngological symptoms. Females were significantly more affected by olfactory and gustatory dysfunctions than males (p=0. Human-to-human transmission is characterized by a troubling exponential rate, which has led to steep curves of onset in many areas [3]. According to the clinical studies from Asia, the most prevalent symptoms consist of fever, cough, dyspnea, sputum production, myalgia, arthralgia, headache, diarrhea, rhinorrhea and sore throat [4,5]. The occurrence of smell dysfunction in viral infections is not new in otolaryngology. Since we focused on the prevalence of olfactory and gustatory disorders, clinical presentation was not considered in as inclusion criteria. The questionnaire has been translated into Spanish, Italian and English by two native speaker otolaryngologists for each language. Olfactory & Gustatory Outcomes the occurrence of olfactory dysfunction has been identified through several questions available in Appendix 1. This is a 7-item patient-reported outcome questionnaire including social, eating, annoyance, and anxiety questions. Each item is rated on a scale of 0-3, with higher scores reflecting better olfactoryspecific QoL. The rest of the olfactory and gustatory questions were based on the smell and taste component of the National Health and Nutrition Examination Survey [10]. This population survey was implemented by the Centers for Disease Control and Prevention to continuously monitor the health of adult citizens in the United States through a nationally representative sample of 5,000 persons yearly [10]. The questions have been chosen to characterize the variation, timing and associated-symptoms of both olfactory and gustatory dysfunctions, and, therefore, they suggest a potential etiology. Note that we assessed the mean recovery time of olfaction through 4 defined propositions: 1-4 days; 5-8 days; 9-14 days and >15 days. Referring to the studies that have demonstrated that the viral load was significantly decreased after 14 days [11], we assessed the short-term olfaction non-recovery rate on patients exhibiting a double criteria: an onset of the infection >14 days before the assessment and the lack of general symptoms at the time of the evaluation. The potential associations between epidemiological, clinical and olfactory and gustatory outcomes have been assessed through cross tab generation between 2 variables (binary or categorical variables) and Chisquare test. The most prevalent comorbidities of patients were allergic rhinitis, asthma, high blood pressure and hypothyroidism (Figure 1). Clinical Outcomes the general symptoms of patients during the infection are described in Figure 2. Cough, myalgia, loss of appetite, diarrhea, fever, headache and asthenia were the most prevalent symptoms, accounting for more than 45% of patients. The otolaryngological symptoms most related to the infection were reported in Table 2. The mean time between the onset of the disease and the assessment of this group of patients was 9. The short-term olfaction recovery rate, which was assessed in 59 clinically cured patients, was 44. The different recovery times of the olfactory function of patients who reported a recovery of the olfactory function are available in Figure 3. In the present study, 76 patients did not suffer from nasal obstruction or rhinorrhea (18. Note that 32 patients did not remember if they had gustatory dysfunction and, therefore, they were not considered for the assessment of the gustatory disorder prevalence. The gustatory dysfunction consisted of reduced/discontinued or distorted ability to taste in 78. The olfactory and gustatory disorders were constant and unchanged over the days in 72. Among the cured patients who had residual olfactory and/or gustatory dysfunction, 53. Olfactory and Gustatory Outcome Associations There was no significant association between comorbidities and the development of olfactory or gustatory dysfunctions. Olfactory dysfunction was not significantly associated with rhinorrhea or nasal obstruction. There was a significant positive association between olfactory and gustatory dysfunctions (p<0. The statistical analysis identified a significant association between the fever and the occurrence of olfactory dysfunction (p=0. The females would be proportionally more affected by olfactory dysfunction compared with males (p<0. The treatments that have been most used for olfactory dysfunction were nasal saline irrigations (16. As a result, the patients were not isolated and the spread of the virus continued. Second, the olfactory dysfunction may appear before, during, or after the general symptoms, with the occurrence of fever being associated with the olfactory dysfunction. In addition to the high prevalence, physicians must keep in mind that olfactory disorder may appear before the rest of the complaints in 11. One of the most important questions from the otolaryngologists concerned the recovery of olfactory and gustatory functions. In the same vein, some patients seemed to recover olfaction, but not taste, and vice versa. Naturally, there are short-term observations and it is reasonable to think that a large number of these patients will recover the olfactory or gustatory functions over the weeks following the disease resolution. Coronavirus has already been identified as a family of viruses that may be associated with anosmia [6]. Moreover, they observed that some patients with normal acoustic rhinometry did not recover their olfaction, suggesting that nasal inflammation and related obstruction were not the only etiological factors underlying the olfactory dysfunction in viral infection. The ability of human coronavirus to invade the olfactory bulb and, therefore, the central nervous system, is most likely a future research path for improving the knowledge about the clinical presentation of patients. From a biomolecular standpoint, viruses could infect peripheral neurons, using the cell machinery of active transport to access the central nervous system [13]. Interestingly, authors demonstrated that the virus antigen was first detected 60 to 66 hours post-infection and was most abundant in the olfactory bulb. Regions of the cortex (piriform and infralimbic cortices), basal ganglia (ventral pallidum and lateral preoptic regions), and midbrain (dorsal raphe) were also strongly infected after the virus had spread [14]; these regions are connected with the olfactory bulb.

Cheap myambutol 400mg with mastercard
Apple-peel atresia is characterized by absence of a vast segment of the small bowel antibiotic for yeast uti 400mg myambutol fast delivery, which can include distal duodenum, the entire jejunum and proximal ileus. The most frequent site of small bowel obstruction is distal ileus (35%), followed by proximal jejunum (30%), distal jejunum (20%), proximal ileus (15%). Anorectal atresia results from abnormal division of the cloaca during the 9th week of development. Prevalence Intestinal obstruction is found in about 1 per 2000 births; in about half of the cases, there is small bowel obstruction and in the other half anorectal atresia. Etiology Although the condition is usually sporadic, in multiple intestinal atresia, familial cases have been described. In contrast with anorectal atresia, associated defects such as genitourinary, vertebral, cardiovascular and gastrointestinal anomalies are found in about 80% of cases. Diagnosis the lumens of the small bowel and colon do not normally exceed 7 mm and 20 mm, respectively. Diagnosis of obstruction is usually made quite late in pregnancy (after 25 weeks), as dilatation of the intestinal lumen is slow and progressive. Jejunal and ileal obstructions are imaged as multiple fluid-filled loops of bowel in the abdomen. If bowel perforation occurs, transient ascites, meconium peritonitis and meconium pseudocysts may ensue. Polyhydramnios (usually after 25 weeks) is common, especially with proximal obstructions. When considering a diagnosis of small bowel obstruction, care should be taken to exclude renal tract abnormalities and other intra-abdominal cysts such as mesenteric, ovarian or duplication cysts. In anorectal atresia, prenatal diagnosis is usually difficult because the proximal bowel may not demonstrate significant dilatation and the amniotic fluid volume is usually normal; occasionally calcified intraluminal meconium in the fetal pelvis may be seen. Prognosis Infants with bowel obstruction typically present in the early neonatal period with symptoms of vomiting and abdominal distention. The prognosis is related to the gestational age at delivery, the presence of associated abnormalities and site of obstruction. In those born after 32 weeks with isolated obstruction requiring resection of only a short segment of bowel, survival is more than 95%. Loss of large segments of bowel can lead to short gut syndrome, which is a lethal condition. It derives from failure of migration of neuroblasts from the neural crest to the bowel segments, which generally occurs between the 6th and 12th weeks of gestation. Another theory suggests that the disease is caused by degeneration of normally migrated neuroblasts during either preor postnatal life. Etiology It is considered to be a sporadic disease, although in about 5% of cases there is a familial inheritance. Diagnosis the aganglionic segment is unable to transmit a peristaltic wave, and therefore meconium accumulates and causes dilatation of the lumen of the bowel. The ultrasound appearance is similar to that of anorectal atresia, when the affected segment is colon or rectum. Polyhydramnios and dilatation of the loops are present in the case of small bowel involvement; on this occasion, it is not different from other types of obstruction. Prognosis Postnatal surgery is aimed at removing the affected segment and this may be a two-stage procedure with temporary colostomy. Bowel perforation usually occurs proximal to some form of obstruction, although this cannot always be demonstrated. Etiology Intestinal stenosis or atresia and meconium ileus account for 65% of the cases. Meconium ileus is the impaction of abnormally thick and sticky meconium in the distal ileum, and, in the majority of cases, this is due to cystic fibrosis. Diagnosis In the typical case, meconium peritonitis is featured by the association of intra-abdominal echogenic area, dilated bowel loops and ascites. The diagnosis should be considered if the fetal bowel is observed to be dilated or whenever an area of fetal intraabdominal hyperechogenicity is detected. The differential diagnosis of hyperechogenic bowel includes: intra-amniotic hemorrhage; early ascites; fetal hypoxia; meconium peritonitis; and cystic fibrosis. The prevalence of cystic fibrosis in fetuses with prenatal diagnosis of intestinal obstruction may be about 10%. Prognosis Meconium peritonitis is associated with a more than 50% mortality in the neonatal period. Hepatic enlargement may also be caused by hemangioma, which is usually hypoechogenic, or hepatoblastoma (the most frequent malignant tumor in fetal life), in which there are areas of calcification. Prevalence Hepatic calcifications are found at mid-trimester ultrasonography in about 1 per 2000 fetuses. Etiology the vast majority of cases are idiopathic but, in a few cases, hepatic calcifications have been found in association with congenital infections and chromosomal abnormalities. Prognosis this depends on the presence of associated infection or chromosomal defects. Renal tract anomalies or dilated bowel are the most common explanations, although cystic structures may arise from the biliary tree, ovaries, mesentery or uterus. The correct diagnosis of these abnormalities may not be possible by ultrasound examination, but the most likely diagnosis is usually suggested by the position of the cyst, its relationship with other structures and the normality of other organs. Choledochal cysts Choledochal cysts represent cystic dilatation of the common biliary duct. Prenatally, the diagnosis may be made ultrasonographically by the demonstration of a cyst in the upper right side of the fetal abdomen. The differential diagnosis includes enteric duplication cyst, liver cysts, situs inversus or duodenal atresia. The absence of polyhydramnios or peristalsis may help to differentiate the condition from bowel disorders. Postnatally, early diagnosis and removal of the cyst may avoid the development of biliary cirrhosis, portal hypertension, calculi formation or adenocarcinoma. Ovarian cysts Ovarian cysts are common and they may be found in up to one-third of newborns at autopsy, although they are usually small and asymptomatic. Fetal ovarian cysts are hormone-sensitive (human chorionic gonadotropin from the placenta) and tend to occur after 25 weeks of gestation; they are more common in diabetic or rhesus isoimmunized mothers as a result of placental hyperplasia. The majority of cysts are benign and resolve spontaneously in the neonatal period. Potential complications include development of ascites, torsion, infarction or rupture. Prenatally, the cysts are usually unilateral and unilocular, although, if the cyst undergoes torsion or hemorrhage, the appearance is complex or solid. Large ovarian cysts can be found in association with polyhydramnios, possibly as a consequence of compression of the bowel. Obstetric management should not be changed, unless an enormous or rapidly enlarging cyst is detected or there is associated polyhydramnios; in these cases, prenatal aspiration may be considered. A difficult differential diagnosis is from hydrometrocolpos, which also presents as a cystic or solid mass arising from the pelvis of a female fetus. Other genitourinary or gastrointestinal anomalies are common and include renal agenesis, polycystic kidneys, esophageal atresia, duodenal atresia and imperforate anus. Mesenteric or omental cysts Mesenteric or omental cysts may represent obstructed lymphatic drainage or lymphatic hamartomas. Antenatally, the diagnosis is suggested by the finding of a multiseptate or unilocular, usually mid-line, cystic lesion of variable size; a solid appearance may be secondary to hemorrhage. Antenatal aspiration may be considered in cases of massive cysts resulting in thoracic compression. Postnatal management is conservative and surgery is reserved for cases with symptoms of bowel obstruction or acute abdominal pain following torsion or hemorrhage into a cyst.